Robust Multi-Omics Prediction for RNA Expression & Protein Surface Levels
Leveraging Single-Cell Multi-Omics Data with Machine Learning to Predict Gene Expression and Protein Levels
Understanding how DNA, RNA, and proteins interact within individual cells can provide important insights into cellular function and disease. Recent advances in single-cell genomics have enabled the measurement of multiple molecular modalities within the same cell. In this project, we developed machine learning models to predict RNA expression from chromatin accessibility data and protein levels from RNA expression in single hematopoietic stem and progenitor cells.
Background
Multi-omics single-cell data provides a unique opportunity to model the complex regulatory relationships between different layers of molecular information. Chromatin accessibility indicates which regions of DNA are available for transcription, directly influencing gene expression. Likewise, RNA levels serve as a template for protein synthesis. By training models to predict across modalities, we can better understand these processes.
Methods
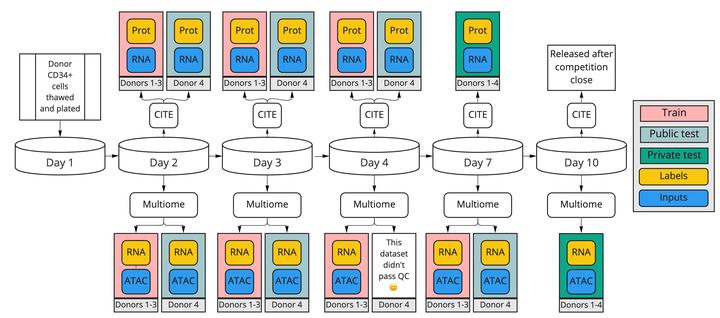
We utilized two large datasets from the Kaggle Competition Open Problems - Multimodal Single-Cell Integration that we already worked on during the Fall semester:
- Multiome: 105,942 cells, 228,942 genomic features, 23,418 RNA targets
- CITEseq: 70,988 cells, 22,050 genes, 38 protein targets
To handle the large data sizes and high sparsity, we converted the data to sparse matrices using scipy.sparse.csr_matrix. This allowed for significant memory optimization, as sparse matrices only store nonzero values.
We then performed extensive preprocessing, including normalization, scaling, and dimensionality reduction using truncated singular value decomposition (SVD). SVD allowed us to extract the most important signals and reduce the data to tractable sizes for modeling.
For each dataset, we compared multiple regression algorithms, including elastic net, LightGBM, and neural networks. Models were trained on early time points and evaluated on their ability to predict unseen future time points.
We performed hyperparameter optimization using RandomizedSearchCV and Bayesian Optimization to find the best model configurations. Regularization and neural architecture tuning were critical to prevent overfitting.
Results
The final models demonstrated excellent predictive performance in cross-validation, with Pearson correlations above 0.9 between predicted and true labels. This suggests chromatin accessibility is highly indicative of gene expression patterns, and RNA levels accurately reflect protein abundance.
Conclusions
This work showed RNA and protein levels can be predicted directly from sequence data alone. Our models leveraged single-cell multi-omics measurements to learn regulatory relationships. The success highlights the wealth of information encoded across modalities. Integrative computational analysis will be critical for maximizing insights. Future work should investigate more modalities and model dynamics over time.
Overall this project demonstrated the power of combining cutting-edge genomics with machine learning to uncover new biology. Predictive modeling across layers of cellular information represents an exciting area for continued methodology development and discovery.